Inter-Clade Recombinant Mpox Virus Detected in England in a Traveller Recently Returned from Asia
Authors:
Steven T. Pullan1, Isobel Everall1, Rebecca Doherty1, Lucy Crossman1, Emma Wise1, Hassan Hartman1, Natalie Groves1, Dominic Worku1, Catherine Houlihan1, Tommy Rampling1, Claire Gordon1, Meera Chand1, Andrew Rambaut2
1 UK Health Security Agency
2 Institute of Ecology and Evolution, University of Edinburgh
A vesicular swab from a traveller returning from a country in Asia tested positive for mpox virus (MPXV) and was initially typed as Clade Ib by qPCR using the G2R-WA, C3L, and C3L flanking region assays [1, 2] at the UKHSA Rare & Imported Pathogens Laboratory.
A near-complete genome (84% consensus coverage) was generated using the ARTIC 2.5 kb tiling amplicon scheme (artic-inrb-mpox/2500/v1.0.0) and Oxford Nanopore sequencing. A consensus sequence was produced via the artic-mpxv-nf v2.0.0 pipeline with Clair3 , using reference PP601219.1 (Clade Ib).
Initial phylogenetic analysis revealed that the consensus sequence was distinct from known Clade Ib sequences, and competitive mapping indicated regions more similar to Clade IIb reference sequences suggesting this was an inter-Clade recombinant.
Additional Testing for Sequence Confirmation
To confirm this unusual finding, amplicon sequencing was repeated on:
- A technical amplification and sequencing repeat from the original extract
- A fresh extract from the same primary sample
- A second, independent swab collected from the patient at the same time
- A VERO E6 cultured isolate derived from the initial swab
Sequences from both swabs and the cultured isolate were identical, supporting the initial observation and demonstrating this is a replication-competent virus.
Recombination Analysis
The putative recombinant sequence was aligned to the Clade Ib reference genome and to a Clade IIb reference genome independently using squirrel [3]. These two reference-aligned genomes were then incorporated into alignments of available genomes from Clade Ib and Clade IIb respectively. For Clade IIb, lineage B.1 and its descendants were excluded to reduce the alignment size and because preliminary analysis showed this lineage was not a constituent part of the recombinant.
The two reference-based alignments (Clade Ib and Clade IIb) were then aligned to each other using consensus alignment in MAFFT [4]. Thus, the putative recombinant appears twice in the final multiple sequence alignment – once optimally aligned to Clade Ib and once to Clade IIb. This allows SNPs that originated in one clade or another to be readily identified. Recombinant tracts were then defined as being between the first SNP unambiguously derived from one clade to the last SNP of that type (Table 1). The regions between these SNPS are ambiguous as to their source but the recombination break points lie in these regions.
Table 1.
Tract
Clade
Start 1
End 1
Length
1
Ib
503
5,365
4,863
2
IIb
5,704
6,112
409
3
Ib
6,205
20,378
14,174
4
IIb
21,508
23,478
1,971
5
Ib
23,831
26,784
2,954
6
IIb
27,550
28,542
993
7
Ib
28,620
29,476
857
8
IIb
29,619
36,963
7,345
9
Ib
37,359
37,812
454
10
IIb
38,084
41,479
3,396
11
Ib
41,851
44,664
2,814
12
IIb
45,300
66,265
20,966
13
Ib
66,576
78,556
11,981
14
IIb
79,321
81,012
1,692
15
Ib
81,669
81,976
308
16
IIb
82,095
87,256
5,162
17
Ib
87,415
87,962
548
18
IIb
88,414
92,269
3,856
19
Ib
92,411
120,396
27,986
20
IIb
121,096
122,971
1,876
21
Ib
123,344
163,282
39,939
22
IIb
163,967
163,967
1
23
Ib
164,058
164,282
225
24
IIb
164,472
164,472
1
25
Ib
164,585
165,302
718
26
IIb
165,399
181,290
15,892
27
Ib
181,399
181,399
1
28
IIb
181,882
186,487
4,606
1 Coordinates are relative to the original sequence.
The Clade Ib-like sequence was constructed by removing all the unambiguously Clade IIb related tracts. Likewise, the Clade IIb-like sequence was constructed by removing Clade Ib tracts. The ambiguous regions between the defined recombinant tracts were left in both sequences. These regions included some mutations unique to the recombinant sequence which may have occurred after the recombination event. These two sequences were then aligned to their respective clade’s genomes sets using squirrel [3] and a phylogenetic tree constructed using IQTree2 [5].
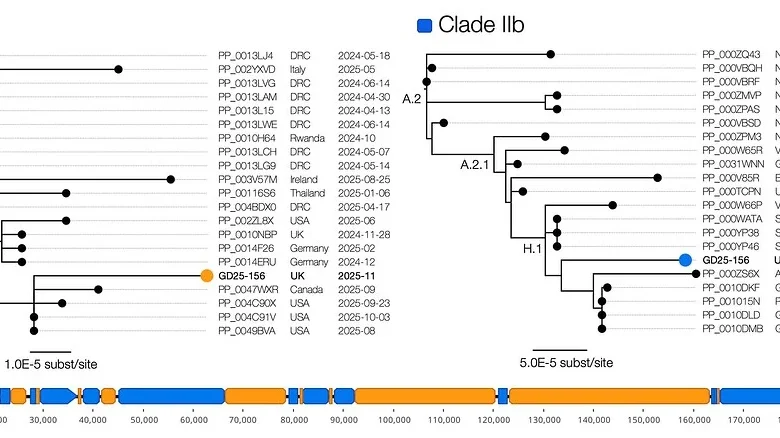
The Clade Ib-only sequence grouped in a cluster of international MPXV genomes including from North America, Europe and Asia and dating from Dec 2024 to Oct 2025 (Figure 1). Given the geographical range and time span, it is likely that this lineage is circulating globally since its emergence in the Democratic Republic of the Congo in late 2023 [6].
The Clade IIb-only sequence was placed in Lineage H.1 (an A.2.1 sublineage) – a lineage that has emerged from Nigeria and is circulating in multiple continents. Note that this lineage is not connected to the B.1 lineage that emerged in 2022 to establish the global epidemic.
Figure 1 | The recombinant structure of the genome comprising 9 Clade Ib related regions (orange) and 8 Clade IIb related regions (blue). The trees show the corresponding phylogenetic relationships for the most closely related Clade Ib genomes (left) and Clade IIb genomes (right).
Discussion
Poxviruses are known to be highly recombinant but co-infections with two different viruses will be rare and in most cases the viruses will have very similar genomes making the resulting recombinants difficult to detect. Even in locations where both clades are circulating and co-infections are possible, most recombinants between divergent viruses will be less fit than the constituent genomes and never observed. It is not possible to determine from a single genome how long this recombinant has been circulating or whether this particular recombinant will have a fitness advantage over currently circulating lineages. However, this event demonstrates that inter-clade recombination can produce viable viruses and highlights the potential for MPXV to continue to evolve as it circulates in the human population. Global genomic surveillance is essential to ensure that any successful variants that emerge are identified and assessed.
Sequence data
The consensus sequence will be available at the European Nucleotide Archive as Assembly: GCA_977880215.1
References
- Li, Y., et al. (2010) Real-time PCR assays for the specific detection of monkeypox virus West African and Congo Basin strain DNA*.* J Virol Methods. 169: 223–227. https://doi.org/10.1016/j.jviromet.2010.07.012
- Schuele, L., et al. (2024) Real-time PCR assay to detect the novel Clade Ib monkeypox virus, September 2023 to May 2024*.* Euro Surveill. 29: 2400486. https://doi.org/10.2807/1560-7917.es.2024.29.32.2400486
- O’Toole, Á., et al. (2025) Human outbreak detection and best practice MPXV analysis and interpretation with squirrel. Virus Evolution. In press.
- Katoh, K., et al. (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform, Nucleic Acids Research. 30: 3059-3066 https://doi.org/10.1093/nar/gkf436
- B.Q. Minh, et al. (2020) IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol., 37:1530-1534. https://doi.org/10.1093/molbev/msaa015
- E.H. Vakaniaki, et al. (2024) Sustained human outbreak of a new MPXV clade I lineage in eastern Democratic Republic of the Congo. Nat Med 30 , 2791–2795. https://doi.org/10.1038/s41591-024-03130-3




